Aminozuurstofwisseling
Aminozuurafbraak
In tegenstelling tot vetzuren (zie vetzuurstofwisseling) en glucose (zie glycogeen metabolisme) kunnen aminozuren niet worden opgeslagen. Aminozuren kunnen ook niet uitgescheiden worden. Een te veel aan aminozuren wordt als brandstof gebruikt. Daartoe wordt de alfa-aminegroep (als NH3) verwijderd en het overblijvende koolstofskelet wordt omgezet zodat het verder gebruikt kan worden in de stofwisseling. Het grootste deel van de aminegroepen van een te veel aan aminozuren worden omgezet in ureum, terwijl hun koolstofskeletten worden omgevormd in acetyl CoA, acetoacetyl CoA, pyruvaat of een tussenproduct van de citroenzuurcyclus. Hieruit kunnen aminozuren omgezet worden tot vetzuren, ketonstoffen en glucose.
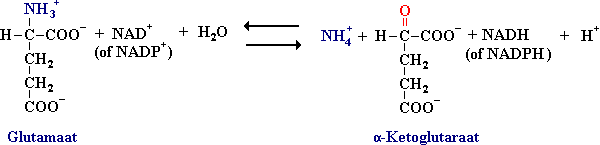
Alfa-aminegroepen worden omgezet in ammoniumionen door deaminering van glutamaat.
De afbraak van aminozuren vindt bij zoogdieren vooral in de lever plaats. Aminetransferasen (transaminasen) katalyseren de overdracht van een aminegroep van een aminozuur naar een ketozuur.
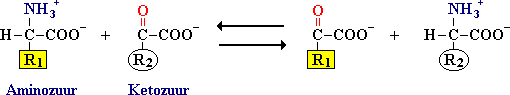
Voorbeelden:
Aspartaat aminetransferase (ASAT = GOT: glutamaat oxalaat transaminase) katalyseert:
aspartaat + alfa-ketoglutaraat ![]() glutamaat + oxaalacetaat.
glutamaat + oxaalacetaat.
Alanine aminetransferase (ALAT = GPT: glutamaat pyruvaat transaminase) katalyseert:
alanine + alfa-ketoglutaraat ![]() glutamaat + pyruvaat
glutamaat + pyruvaat
Glutamaatdehydrogenase katalyseert de reactie waarbij ammoniumionen (NH4+) worden gevormd uit glutamaat met NAD+ of NADP+ als het oxiderende coënzymen. Het enzym wordt geremd door GTP en ATP en geactiveerd door GDP en ADP. Een verslechtering van de energiestatus in de cel versnelt dus de oxidatie van aminozuren.
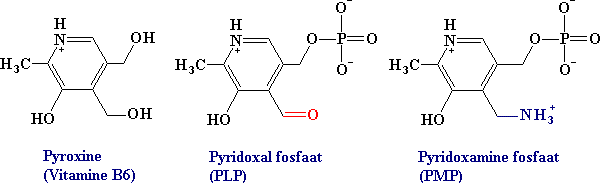
PLP is de hulp groep voor aminotransferasen
Pyridoxalfosfaat vormt Schiff basen als tussenproducten met aminotransferasen. Pyridoxalfosfaat (PLP) os de hulp groep voor alle aminetransferasen. PLP is een omzettingsproduct van pyridoxine (vitamine B6). Tijdens de transaminering wordt PLP tijdelijk omgezet in pyridoxaminefosfaat (PMP).
In de afwezigheid van substraat is de aldehydegroep van PLP gebonden aan de aminegroep (een Schiff base binding) van een bepaald lysineresidu op de actieve plaats van het aminetransferase. Bij aanwezigheid van een aminozuursubstraat wordt een nieuw Schiff base binding gevormd. De alfa-aminegroep van het aminozuur neemt de plaats in van de epsilon-aminegroep van het lysineresidu op de actieve plaats. Met andere woorden: een intern aldimine wordt een extern aldimine.
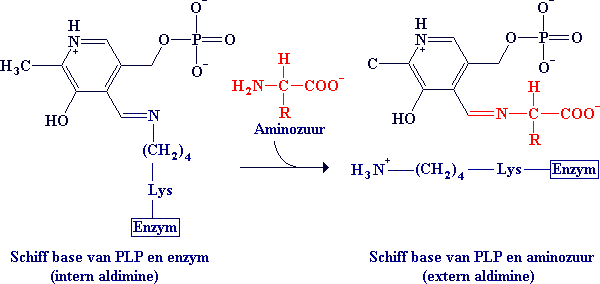
Het extern aldimine verliest een proton van zijn alfa-koolstofatoom onder vorming van een quinon-achtig tussenproduct. Protonering levert een ketimine op met een dubbele binding tussen het N-atoom en het C-atoom van het substraat. In het aldimine daarentegen was er een dubbele binding tussen het N-atoom en het carbonyl koolstofatoom van PLP. Het ketimine wordt dan gehydrolyseerd in een alfa-ketozuur en PMP.
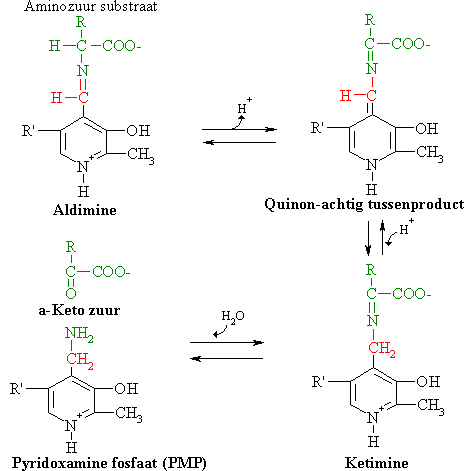
Een ander (tweede) alfa-ketozuur kan de omgekeerde reacties ondergaan onder vorming van een ander (tweede) aminozuur.
De totaalreactie is dan:aminozuur(1) + alfa-ketozuur(2)
De uitscheiding van amoniumionen
Een deel van het NH4+ dat gevormd wordt bij de afbraak van aminozuren wordt gebruikt voor de biosynthese van stikstofverbindingen. Bij de meeste op het land levende gewervelde dieren (inclusief de mens) wordt de overmaat NH4+ omgezet in ureum en in die vorm uitgescheiden. Meer hierover is de lezen op de pagina ureum cyclus. Bij vogels en op het land levende reptielen wordt NH4+ voor de uitscheiding omgezet in urinezuur. Bij veel in het water levende dieren wordt NH4+ zelf uitgescheiden in het water.
Bestemming van afgebroken aminozuren in de stofwisseling
De koolstofatomen van afgebroken aminozuren komen terug in belangrijke tussenproducten van de stofwisseling. De strategie van de aminozuurafbraak is de vorming van belangrijke tussenproducten die omgezet kunnen worden in glucose of geoxideerd kunnen worden door de citroenzuurcyclus. De koolstofskeletten van de twintig aminozuren worden teruggebracht tot slechts zeven moleculen: pyruvaat, acetyl CoA, acetoacetyl CoA, alfa-ketoglutaraat, succinyl CoA, fumaraat en oxaalacetaat. Aminozuren die worden afgebroken tot acetyl CoA of acetoacetyl CoA worden ketogene aminozuren genoemd omdat zij omgezet kunnen worden in ketonstoffen. De aminozuren die worden omgezet in de overige van de zeven moleculen worden glucogene aminozuren genoemd omdat zij omgezet kunnen worden in fosfo-enolpyruvaat en vandaar uit in glucose (via de gluconeogenese).
Bestemmingen van de koolstofketens van aminozuren. Glucogene aminozuren staan in een rood kader, de ketogene aminozuren in een geel kader. Sommige aminozuren (ile, try, phe, tyr) zijn zowel glucogeen als ketogeen.
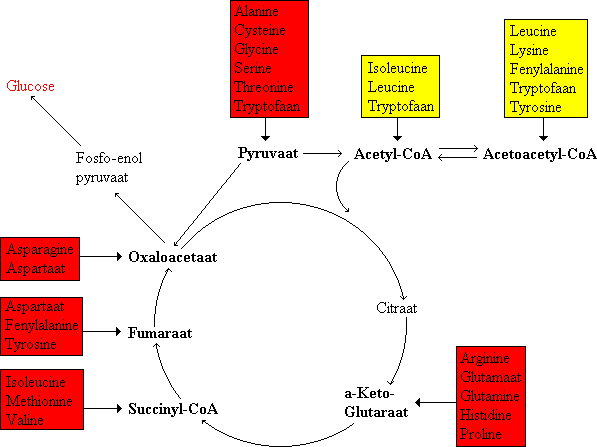
De C3 familie: alanine, serine en cysteine worden omgezet in pyruvaat. Pyruvaat is het toegangspunt voor alanine, serine, cyseine, glycine, threonine en tryptofaan. Cysteine kan door verschillende paden omgezet worden in pyruvaat waarbij het zwavelatoom terugkomt in H2S, SO32-, of SCN-. Ook koolstofatomen van de aminozuren glycine, threonine en tryptofaan kunnen terugkomen in pyruvaat.
De C4 familie: aspartaat en asparagine worden omgezet in cxaalacetaat. Aspartaat kan ook door de ureumcyclus worden omgezet in fumaraat. Fumaraat is een toegangspunt voor de helft van de koolstofatomen van tyrosine en fenylalanine.
De C5 familie: glutamine, proline, arginine en histidine worden omgezet in alfa-ketoglutaraat via glutamaat. Alfa-ketoglutaraat is het toegangspunt van glutamine, proline, arginine en histidine die eerst worden omgezet in glutamaat.
Barnsteenzuur CoA is een toegangspunt voor enkele apolaire aminozuren. Omzetting van methionine, isoleucine en valine via propionyl CoA in barnsteenzuur CoA. De route van propionyl CoA naar barnsteenzuur CoA wordt ook doorlopen in de oxidatie van vetzuren met een oneven aantal koolstofatomen. Vetzuren met een oneven aantal koolstofatomen zijn dus gedeeltelijk glucogeen; drie van hun koolstofatomen kunnen terugkomen in glucose.
Afbraak van fenylalanine en tyrosine in acetylacetaat en fumaraat
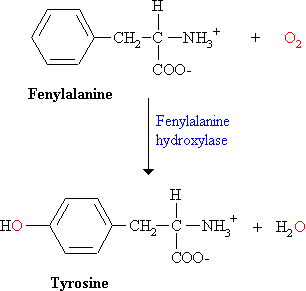
De eerste stap is de hydroxylering van fenylalanine tot tyrosine door het enzym fenylalanine hydroxylase. Dit enzym wordt een monooxygenase of mixed-function oxygenase genoemd omdat één atoom van O2 in het product (tyrosine) terechtkomt en de ander in H2O.
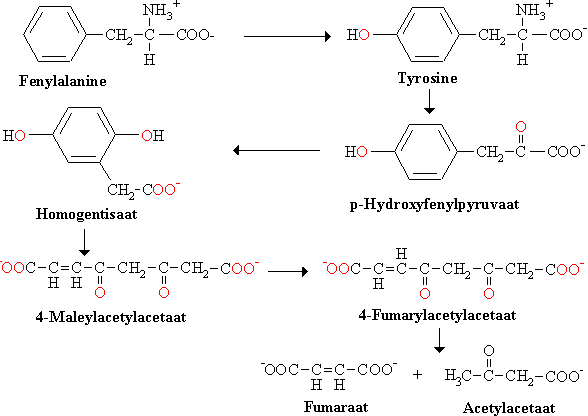
De tweede stap is de transaminering van tyrosine in p-hydroxyfenylpyruvaat. Dit alfa-ketozuur reageert met O2 onder vorming van homogentisaat (het zuurrest van homogentisinezuur). Het enzym p-hydroxyfenylpyruvaat hydroxylase is een dioxygenase omdat beide atomen van O2 in het product worden ingebouwd (één in de ring en één in de carboxylgroep).
De aromatische ring wordt vervolgens opengebroken door O2 onder vorming van 4-maleylacetylacetaat. Deze reactie wordt gekatalyseerd door een ander dioxygenase enzym.
4-Maleylacetylacetaat wordt dan geisomeriseerd tot 4-fumarylacetylacetaat. Tenslotte wordt 4-fumarylacetylacetaat gehydrolyseerd in fumaraat en acetylacetaat. Zie de onderstaande figuur voor het pad van de afbraak van fenylalanine en tyrosine.
Een blokkade in de afbraak van fenylalanine kan tot geestelijke achterstand leiden
De afwezigheid of onvoldoende activiteit (deficiëntie) van fenylalaninehydroxylase (of zijn cofactor tetrahydrobiopterine) veroorzaakt een ophoping van fenylalanine in alle lichaamsvloeistoffen en daardoor een uitscheiding in de urine van het omzettingsproduct fenylpyruvaat (fenylketon).
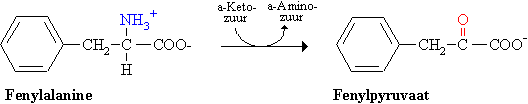
De biochemische achtergrond van de mentale retardatie is nog een raadsel. Een vroege diagnose is essentieel en wordt bereikt door een screening van pasgeborenen. In het bloed, verkregen door middel van een hielprikje, wordt de concentratie fenylalanine bepaald. De frequentie van fenylketonurie is 1 op 20.000 pasgeborenen. De ziekte wordt autosomaal recessief overgeërfd. De geestelijke achterstand (mentale retardatie) wordt voorkomen door een dieet met weinig fenylalanine. Daartoe worden eiwitten met een laag gehalte aan fenylalanine zoals het melkeiwit caseïne gehydrolyseerd. Uit het hydrolysaat wordt fenylalanine verwijderd door adsorptie.